
Michał Krzysztof Jastrzębski

PC13 – Michał Jastrzębski
Chair and Department of Synthesis and Chemical Technology of Pharmaceutical Substances, Faculty of Pharmacy, Medical University of Lublin, 4A Chodźki St., PL-20093 Lublin, Poland
michal.jastrz1998@gmail.com
| Synthesis, molecular modeling and assessment of anticancer activity of new CYP17A1 inhibitors of the D2AAK1M series |
| Michał K. Jastrzębski1, Tomasz M. Wróbel1, Agnieszka Korga-Plewko2, Magdalena Iwan3, Joanna Kubik2 and Agnieszka A. Kaczor1 1 Chair and Department of Synthesis and Chemical Technology of Pharmaceutical Substances, Faculty of Pharmacy, Medical University of Lublin, 4A Chodźki St., PL-20093 Lublin, Poland 2 Independent Medical Biology Unit, Medical University of Lublin, 8b Jaczewski Street, 20-090 Lublin, Poland 3 Department of Toxicology, Faculty of Pharmacy, Medical University of Lublin, Chodźki 8, 20-093 Lublin, Poland. |
| Abstract Prostate cancer (PCa) is the second leading cause of cancer-related mortality in men. A key therapeutic target in castration-resistant prostate cancer (CRPC) is the CYP17A1 enzyme, which catalyzes androgen biosynthesis. Abiraterone, currently used in clinical practice, possesses a steroidal scaffold, which limits its selectivity and leads to adverse effects. This work presents the development of a new series of non-steroidal inhibitors, D2AAK1M (Fig. 1), representing an evolution of pyridine-indole hybrids [1,2]. 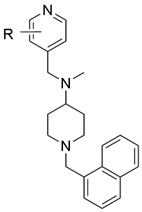 Figure 1. Structure of D2AAK1M derivatives. The designed compounds were based on a 4-substituted pyridine scaffold (the heme-binding group) connected via an aliphatic linker to a naphthalene moiety. PASS analysis demonstrated high probability of both antitumor activity and selective inhibition of steroidogenic enzymes. Molecular docking studies (SeeSAR) confirmed that D2AAK1M derivatives occupy the same binding pocket as abiraterone. The molecules adopt an optimal pose: the scaffold is oriented at a 60° angle relative to the heme, with the pyridine nitrogen atom coordinating the iron at an approximately perpendicular angle. In vitro experiments using PCa cell lines (LNCaP, DU-145, PC3) and BJ fibroblasts demonstrated that certain compounds selectively influenced cell viability. These agents significantly reduced the viability of the LNCaP (AR+) cell line at 100 µM. Simultaneously, they remained inactive against AR-negative lines (Du-145, PC3) and exhibited no toxicity toward normal cells. Selective effects in AR+ support the hypothesis of a mechanism based on the inhibition of androgen production. The D2AAK1M series represents a promising starting point for the development of novel anticancer drugs with an improved safety profile. |
| References [1] Wróbel, T.M.; Grudzińska, A.; Yakubu, J.; et al. Pyridine indole hybrids as novel potent CYP17A1 inhibitors. J. Enzyme Inhib. Med. Chem. 2025, 40(1), 2463014. [2] Wróbel, T. M.; Jørgensen, F. S.; Pandey, A. V.; et al. Non-steroidal CYP17A1 Inhibitors: Discovery and Assessment. J. Med. Chem. 2023, 66 (10), 6542–6566. |